SR69
AR Stargardt Disease (IIA)
Male
Male
SR69
AR Stargardt Disease (IIA)
Male
Male
History
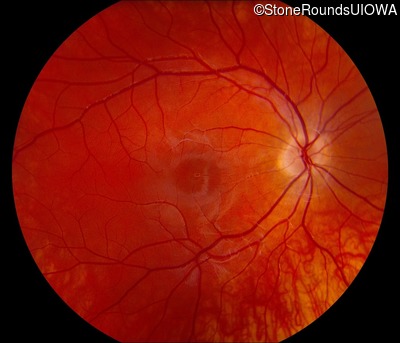
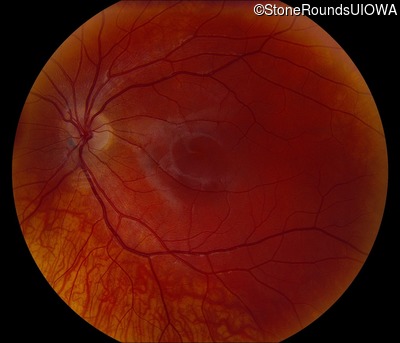
This 16 year old male first noticed some reduction in visual acuity in the right eye more than the left at age 13. His visual acuity had been previously normal. He is now experiencing some photophobia.
| Color Vision: | 12/14 color plates OU |
|---|---|
| Refraction OD: | -2.25 sphere |
| Refraction OS: | -1.00 +0.25 x 085 |
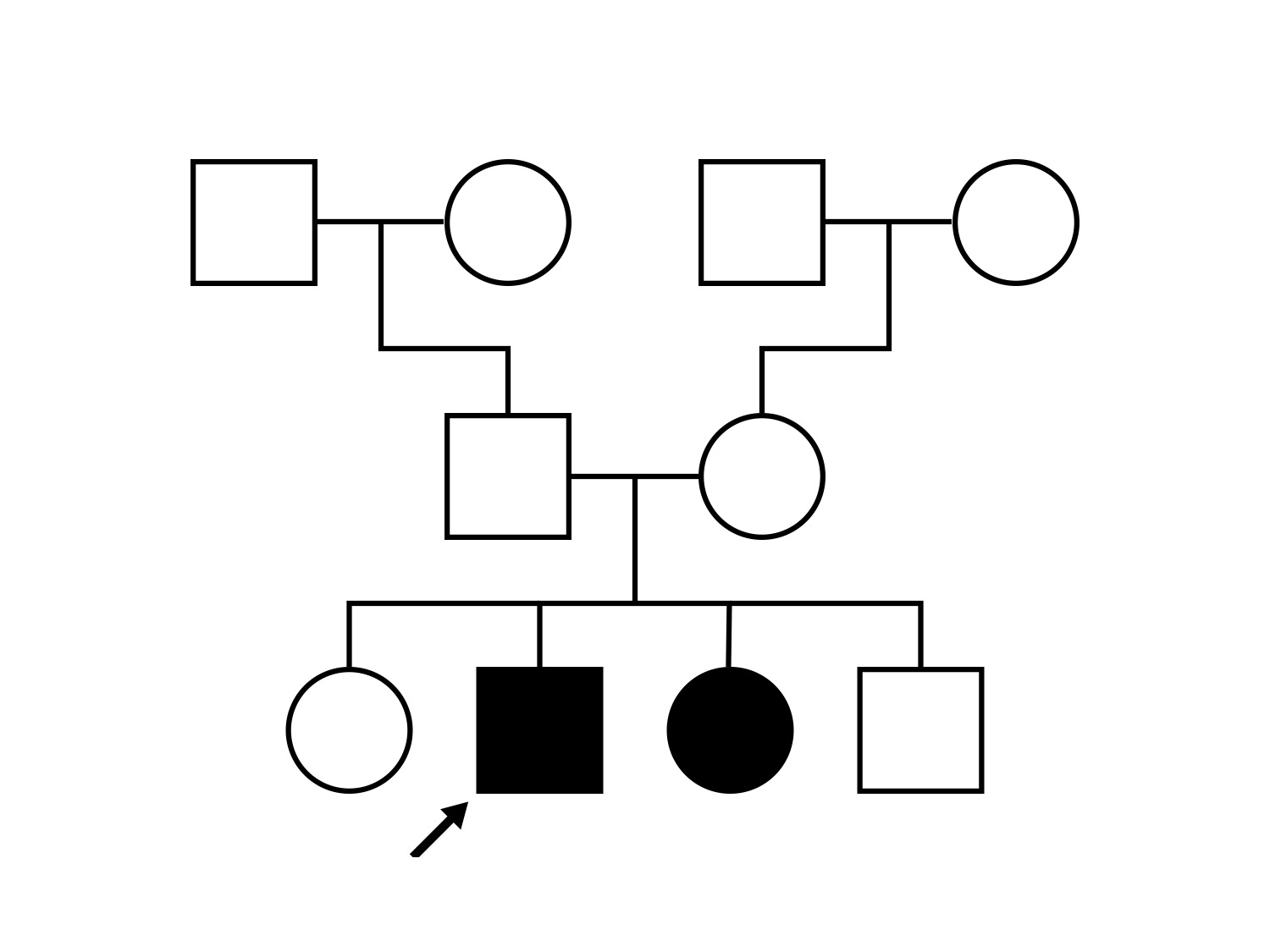
Pedigree
Teaching Points
The clinical features favoring the diagnosis of ABCA4-associated autosomal recessive Stargardt disease include: loss of acuity in the second decade, foveal photoreceptor loss on OCT, a vermilion appearance of the posterior pole of his fundi, a similarly affected sister and normally sighted parents.
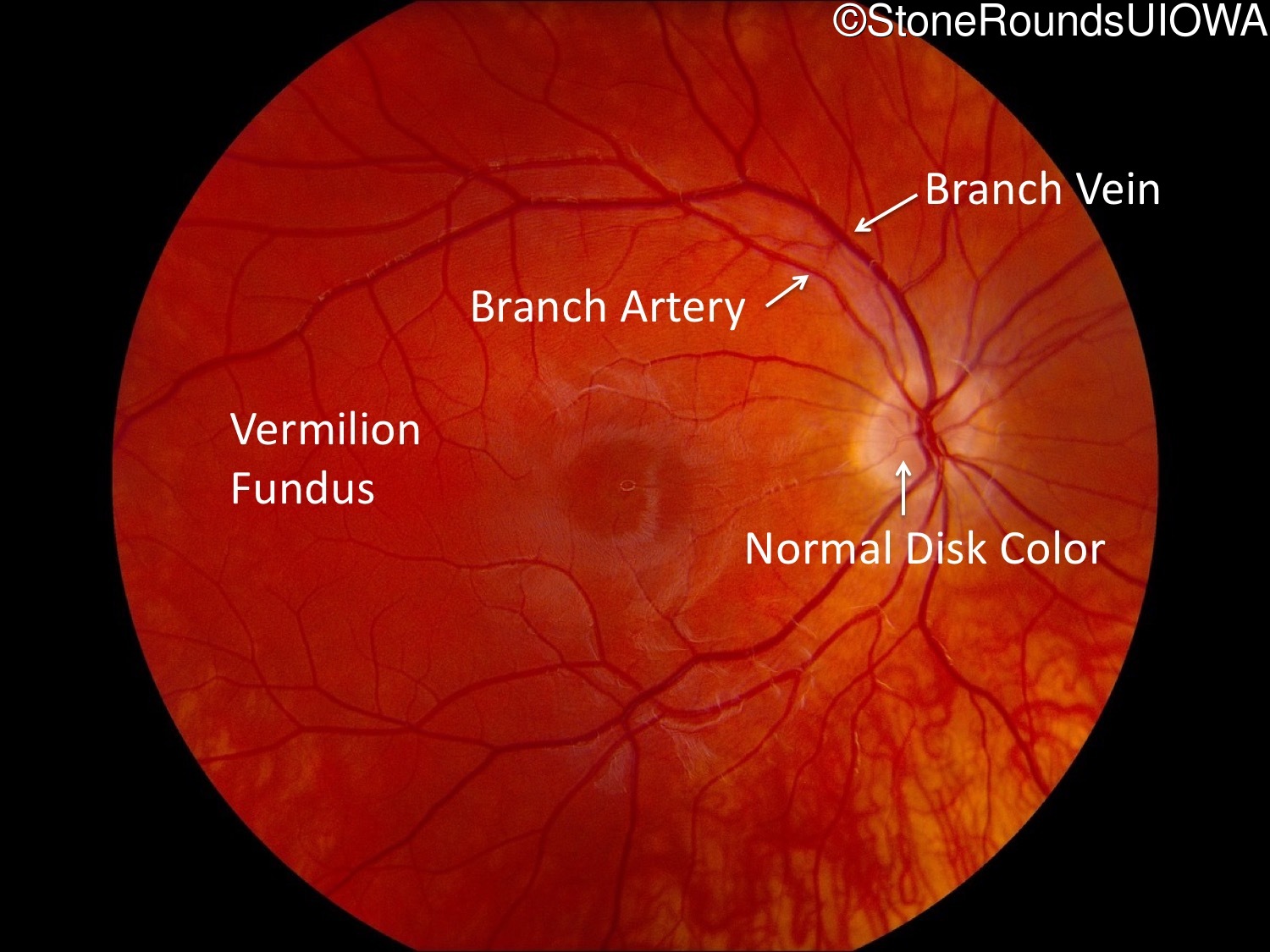
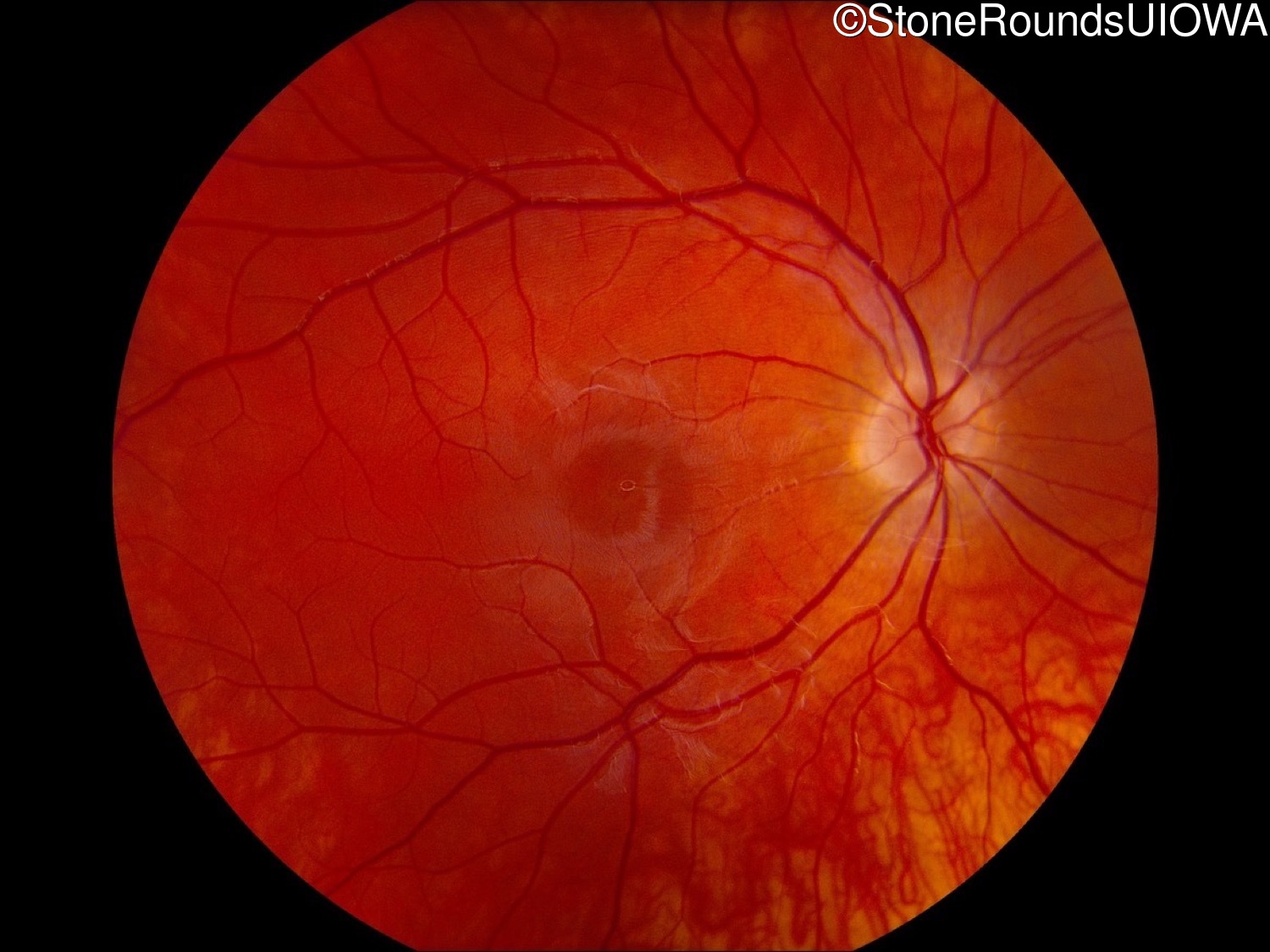
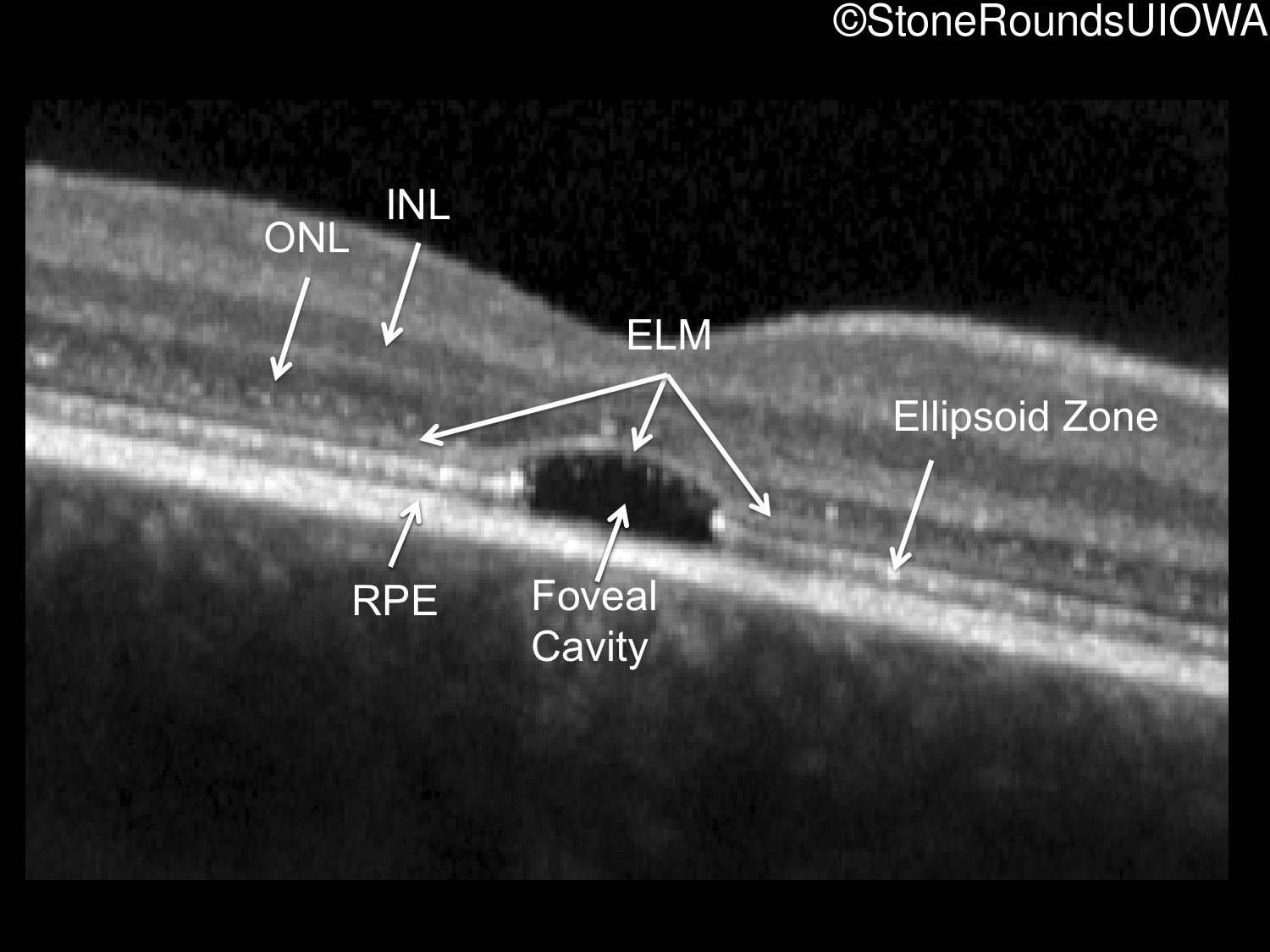
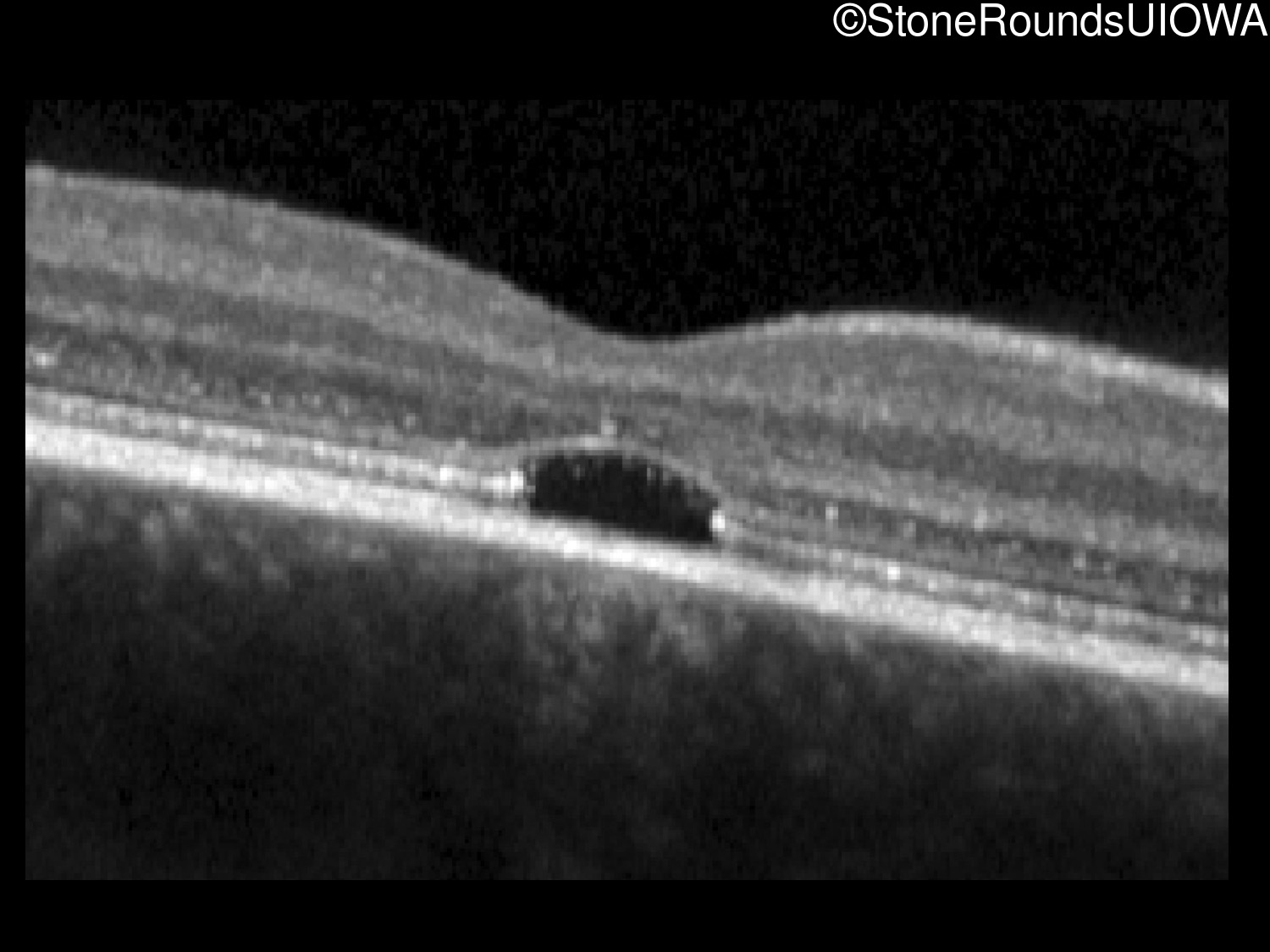
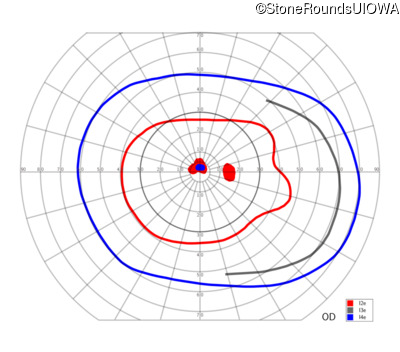
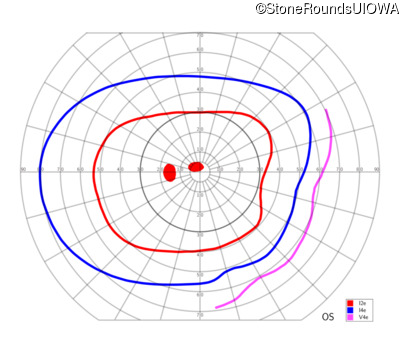
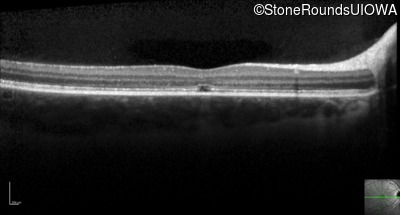
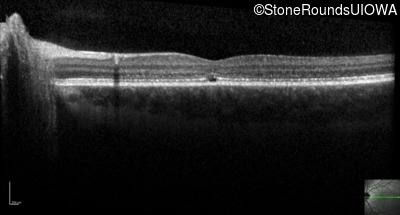
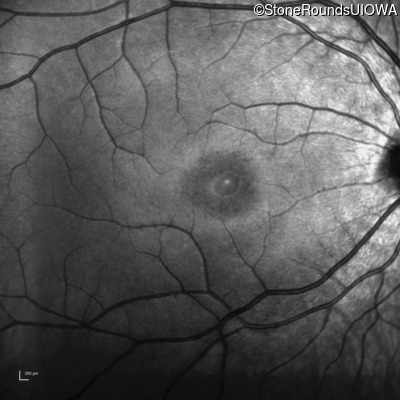
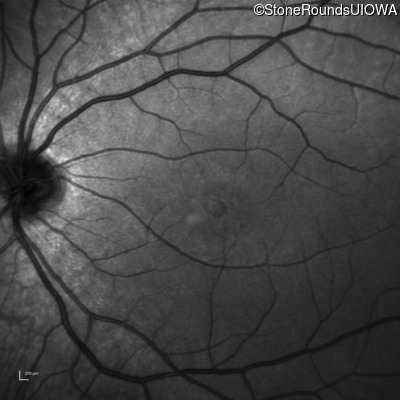
One can usually tell which of the three main branches of inherited retinal disease a patient has just by knowing the age and the visual acuity and looking at the fundus photos. In this case, however, the fundus and disk are pretty normal looking. There are two subtle findings that could be normal but are noteworthy given the poor acuity. First, the retinal branch arterioles seem slightly narrow for a 16 year old. Second, the entire posterior pole is blood red suggesting that the patient is has very little melanin in his RPE and choroid. Despite this, there is little choroidal detail visible in the macula. This is sometimes called a "vermilion fundus" and can result from a lot of A2E accumulation in the RPE blocking one's view of the large choroidal vessels. The OCT reveals a loss of foveal photoreceptors and a fluid filled space where these photoreceptors should be. In addition, there is a subtly motheaten appearance of the ellipsoid zone throughout the macula, a loss of the outer nuclear layer throughout the macula, and a thickening of the external limiting membrane throughout the macula. These things suggest cone-selective photoreceptor loss, which is found in achromatopsia, cone dystrophy,RP1L1-associated occult macular dystrophy, and some patients with Stargardt disease. The latter is the most common single entity seen in an inherited retinal disease clinic (18%) and so would be at the top of the list at this point in the analysis. The subjective report of photophobia is supportive of a cone selective disease [because the cones are supposed to inhibit the rods in bright light and, when they are selectively lost, give rise to photophobia]. Two things argue convincingly against achromatopsia: first, the onset of symptoms in the second decade of life and the ability to see 12/14 pseudoisochromatic plates. However, the latter would be pretty unusual for a Stargardt patient with 20/160 acuity too, so I am not sure that the color plates are terribly helpful here. The absence of an affected parent or grandparent argue against a dominant disease while his similarly affected sister strengthens our belief that this is a heritable condition and suggests that it is autosomal recessive (still good for AR Stargardt). The Goldmann visual field is also much more characteristic of a patient with Stargardt disease and a bad foveal factor (see Schindler, et al., 2010) than it is achromatopsia. In the latter condition, the I2e isopter is often completely lost because of the loss of sensitivity associated with loss of one entire photoreceptor class. The slightly myopic refraction is also consistent with Stargardt disease. Patients with this condition are rarely hyperopic.
Diagnosis & molecular findings
| Disease | Gene | Allele 1 variant(s) | Allele 2 variant(s) | Inheritance mode |
|---|---|---|---|---|
| AR Stargardt Disease | ABCA4 | Gly1961Glu GGA>GAA | Asn1442Lys AAC>AAA | AR |